Le cheveu sans racine figure parmi les traces biologiques que l’on peut découvrir sur une scène de crime, et parmi les plus difficiles à exploiter dans les laboratoires de la police scientifique. Dépourvu de racine, il livre rarement un profil exploitable par les méthodes classiques et reste le plus souvent cantonné à l’analyse d’ADN mitochondrial, peu discriminante. Cependant, une étude parue dans Genome Biology en février 2026, signée par l’équipe de Joshua Kapp et Richard Green (University of California Santa Cruz et Astrea Forensics), offre désormais de nouvelles perspectives. En transposant les outils mis au point pour séquencer l’ADN ancien ou dégradé, les auteurs montrent que quelques centimètres de tige pilaire suffisent à établir un profil génétique classique exploitable, permettant de rapprocher ou d’écarter un individu avec un poids statistique élevé. L’enjeu concerne aussi bien les affaires criminelles classiques – notamment les infractions à caractère sexuel, lorsque des cheveux ou des poils pubiens sont retrouvés sur la victime – que les affaires non élucidées (cold cases) ou encore l’identification de restes humains.
Pourquoi le cheveu sans racine échappait à l’analyse ADN nucléaire
L’identification par génétique forensique repose le plus souvent sur l’analyse de marqueurs STR (Short Tandem Repeat), également appelées microsatellites. Il s’agit de courtes séquences d’ADN qui se répètent, et dont le nombre de répétitions varie d’un individu à l’autre. En analysant plusieurs dizaines de ces zones, il est possible d’établir une sorte de « code-barres » génétique propre à chaque personne. Le profil obtenu alimente les bases de données nationales, comme le FNAEG (Fichier National Automatisé des Empreintes Génétiques) en France ou le CODIS aux États-Unis. Cette approche est rapide, robuste et très discriminante, mais elle exige un ADN relativement bien conservé, composé de fragments suffisamment longs (soit plusieurs centaines de nucléotides). Or la tige du cheveu (sans bulbe) ne contient qu’un ADN nucléaire massivement dégradé. L’étude de Kapp et ses collègues (2026) confirme à ce propos que la quasi-totalité des fragments mesurent moins de cent nucléotides, soit bien en deçà de la longueur nécessaire pour réaliser une analyse STR classique. Cette absence d’ADN nucléaire exploitable ne résulte pas d’une dégradation progressive de la trace au fil du temps, mais d’un processus biologique propre à la formation du cheveu. Lors de la kératinisation, c’est-à-dire lorsque les cellules de la tige durcissent et se chargent en kératine, une endonucléase nommée DNase1L2, découpe l’ADN en très petits fragments. Ces fragments résiduels restent ensuite piégés dans la tige, comparables à des courts fragments d’ADN qui circulent librement dans le plasma sanguin. Leur taille très réduite explique ainsi pourquoi l’analyse génétique classique du cheveu sans bulbe était jusqu’ici difficile, voire impossible, et pourquoi les analyses se limitaient le plus souvent à l’ADN mitochondrial.
Les outils de l’analyse d’ADN ancien au service de l’enquête judiciaire
La solution retenue par Kapp et al. (2026) ne consiste pas à améliorer les méthodes d’amplification classiques existantes, mais à adopter une approche différente fondée sur le séquençage massif de l’ADN. Les auteurs appliquent ainsi un protocole optimisé pour l’ADN très dégradé, directement issu de la paléogénomique. Il s’agit de la discipline qui a permis, dès 2010, de reconstituer le génome complet d’un individu ayant vécu il y a près de 4 000 ans à partir d’une simple mèche de cheveux conservée dans les collections d’un musée à Copenhague (Rasmussen et al., 2010).
Chaque cheveu est d’abord nettoyé en surface à l’aide d’une solution diluée d’hypochlorite de sodium, afin d’éliminer l’ADN exogène et les contaminations microbiennes. Cette étape constitue un avantage par rapport à l’os ou à la dent, dont la décontamination est plus complexe. L’ADN est ensuite extrait puis préparé selon une méthode conçue pour conserver les fragments les plus courts, sans les fragmenter davantage. Ces fragments sont enfin analysés par séquençage à haut débit, à raison d’environ 300 millions de lectures par cheveu. Point important pour les laboratoires, l’ensemble du protocole a été pensé pour pouvoir être automatisé, condition essentielle à une éventuelle utilisation en routine. Détail important pour les laboratoires, l’ensemble du flux de travail a été conçu pour l’automatisation, condition d’un éventuel passage en routine.
De quelques centimètres de cheveu à un profil génétique exploitable
À partir de quelques centimètres seulement (cinq centimètres de cheveu ou trois centimètres de poil pubien), les auteurs parviennent à obtenir suffisamment d’ADN nucléaire pour analyser le génome. Plus particulièrement Kapp et al. (2026) atteignent une couverture moyenne du génome de 2,18 fois pour les cheveux et 2,64 fois pour les poils pubiens. La couverture mesure le nombre moyen de fois où chaque position du génome est lue. Une valeur de « 2 » signifie que chaque position a été observée environ deux fois en moyenne. L’essentiel de l’ADN humain récupéré est nucléaire, la part mitochondriale restant minoritaire, autour de 3%. Cette quantité, modeste en apparence, suffit à reconstruire un profil de polymorphismes nucléotidiques, les SNP (Single Nucleotide Polymorphism), ces positions du génome où un seul nucléotide varie d’un individu à l’autre. Comme la couverture demeure faible, les positions non lues sont complétées par imputation, une inférence statistique qui s’appuie sur un panel de référence de génomes connus, ici le projet 1000 Génomes, pour déduire les génotypes manquants à partir des variants voisins. La détermination du sexe est immédiate puisqu’elle découle du rapport entre les lectures portées par le chromosome X et celles portées par un autosome de taille comparable. Pour les 77 cheveux dont la couverture moyenne dépasse une fois, la concordance des génotypes avec l’ADN de référence prélevé dans la salive du même donneur dépasse 99,4%.
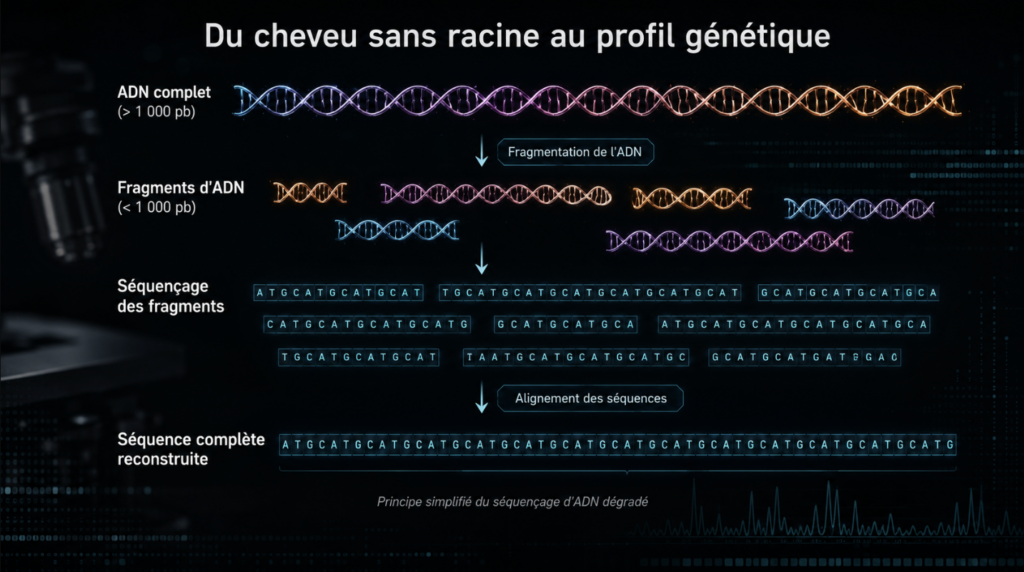
Principe du séquençage par fragments (shotgun sequencing). L’ADN, qu’il soit naturellement dégradé ou volontairement fragmenté, se présente sous la forme de courts segments (ici < 1 000 paires de bases). Chaque fragment est séquencé individuellement, puis les lectures obtenues sont alignées par chevauchement de leurs régions communes. Cet assemblage permet de reconstituer, de proche en proche, la séquence complète de la molécule d’origine — y compris lorsque l’ADN de départ est trop court et trop abîmé pour les méthodes d’amplification classiques. Crédit : Forenseek
Identifier ou écarter une personne : l’approche statistique
L’apport décisif de l’étude de Kapp et al. (2026) tient à la fiabilité de la méthode proposée. Les auteurs ont comparé l’ADN extrait de 80 cheveux avec des profils génétiques de référence établis à partir de la salive de 50 personnes. Ils s’attendaient ainsi à 80 comparaisons positives, lorsque le cheveu et la salive provenaient de la même source, et à près de 4 000 comparaisons négatives, lorsque les échantillons provenaient de sources différentes. Pour chaque comparaison, un outil calcule un rapport de vraisemblance, c’est-à-dire le rapport entre deux probabilités : celle d’observer l’ADN du cheveu si celui-ci provient de la personne comparée, et celle de l’observer s’il provient d’un individu inconnu et non apparenté. Sur l’ensemble des tests, le calcul a fourni le résultat attendu, « identification » ou « exclusion », sans la moindre erreur. Le résultat reste valable même lorsque la quantité d’informations issues du cheveu est volontairement réduite à moins de 0,2 fois de couverture, ce qui correspond à une trace très pauvre en ADN. Les auteurs relèvent que les comparaisons négatives (lorsque le cheveu analysé ne provient pas de la personne de référence) permettent de conclure à une exclusion de manière particulièrement robuste. Les comparaisons positives (lorsque le cheveu et l’échantillon de référence proviennent de la même personne) permettent quant à elles d’établir un rapprochement, mais avec un résultat statistiquement moins marqué. Cette asymétrie permet une véritable prudence en contexte judiciaire, puisqu’elle limite le risque d’attribuer à tort une trace génétique pauvre à une personne. Concrètement, un seul cheveu peut désormais identifier une personne, et non plus seulement fournir un renseignement d’orientation.
Une vraie avancée pour les affaires non élucidées
Les conséquences de cette étude pour les enquêtes judiciaires sont multiples et notables. Les scènes de crime et certains scellés judiciaires (notamment les vêtements et linges de lit) regorgent souvent de cheveux, y compris quand les autres traces font défaut. De nombreux dossiers non résolus (cold cases) conservent par ailleurs des cheveux prélevés pour des examens morphologiques, jusqu’ici considérés comme inexploitables pour l’analyse de l’ADN nucléaire. Le profil SNP dense obtenu grâce à la méthode développée par Kapp et al. (2026) ouvre la voie à la généalogie génétique investigative, qui consiste à rechercher des parents éloignés d’un individu dans les bases de données de tests génétiques grand public, méthode à l’origine de la résolution de centaines d’affaires aux États-Unis. Une distinction importante s’impose toutefois pour le lecteur français. La recherche en parentèle, légalisée par la loi du 3 juin 2016 et codifiée à l’article 706-56-1-1 du code de procédure pénale, est autorisée. Elle permet d’interroger le FNAEG, sur la base de profils STR, pour repérer un proche parent déjà connu des services judiciaires et déjà fiché. La généalogie génétique investigative constitue une démarche différente, puisqu’elle repose sur des profils SNP et sur l’exploitation de bases de données commerciales étrangères. Cette pratique est pour l’heure interdite, le recours aux tests récréatifs étant prohibé en France et passible d’une amende. Le projet de loi dit SURE, porté par le garde des Sceaux, prévoit de l’autoriser dans son article 3, seulement pour les crimes les plus graves et sous le contrôle du juge. Adopté par le Sénat en avril 2026, le texte est en cours d’examen à l’Assemblée nationale et nourrit un vif débat sur le consentement, la fiabilité des bases étrangères et la protection des données personnelles.
Au sein de Forenseek, nous formulons toutefois une réserve quant à la portée de cette étude. En effet, plusieurs auteurs de cette étude sont liés à la société qui développe et commercialise la technologie. De nouvelles études scientifiques et une validation indépendante par des laboratoires accrédités reste à mener avant tout usage dans le cadre judiciaire. Notons que le cheveu peut également être utilisée pour une analyse protéomique à des fins d’identification.
Sources :
- Kapp J. D., Wanket C., Nguyen R. et al. Rootless hair as a reliable source of forensic genetic information. Genome Biology, 2026, 27:104. disponible ici : https://pubmed.ncbi.nlm.nih.gov/41703596/
- Brandhagen M. D., Loreille O., Irwin J. A. Fragmented nuclear DNA is the predominant genetic material in human hair shafts. Genes, 2018.
- Fischer H. et al. DNase1L2 degrades nuclear DNA during corneocyte formation. Journal of Investigative Dermatology, 2007.
- Nguyen R., Kapp J. D., Sacco S. et al. A computational approach for positive genetic identification and relatedness detection from low-coverage shotgun sequencing data. Journal of Heredity, 2023.
- National Institute of Justice. Nuclear DNA from Rootless Hair for Forensic Purposes. Project summary, 2024.
- Code de procédure pénale, articles 706-54 (FNAEG) et 706-56-1-1 (recherche en parentèle) ; loi n° 98-468 du 17 juin 1998 ; loi n° 2016-731 du 3 juin 2016.
- Code civil, article 16-10 (examen des caractéristiques génétiques).
- Projet de loi relatif à la justice criminelle et au respect des victimes, dit SURE, article 3 ; dossiers législatifs du Sénat et de l’Assemblée nationale, 2026.
